How GLP-1 Medications Work
Summary: GLP-1 receptor agonists copy a gut hormone that boosts insulin only when blood sugar is high, blocks glucagon, slows the stomach, and quiets appetite signals in the brain. That four-part mechanism is why drugs like Ozempic, Wegovy, Mounjaro, and Zepbound lower A1C by roughly 1 to 2 percentage points and produce 10 to 20 percent body-weight loss in clinical trials.
This content is for informational purposes only and is not medical advice. Always consult a qualified healthcare provider before starting, changing, or stopping any medication.
What GLP-1 actually is
Glucagon-like peptide-1 is a 30-amino-acid hormone made by L-cells in the lining of your small intestine and colon. When food (especially carbohydrates and fats) reaches the gut, those L-cells release GLP-1 into the bloodstream within minutes. [1] The peptide is cleaved from a larger precursor called proglucagon, the same gene product that yields glucagon in pancreatic alpha cells and a separate pool of GLP-1 in a small population of brainstem neurons in the nucleus tractus solitarius. The fact that one gene serves three different secretory tissues, with three different products, is part of why the GLP-1 system reaches into the pancreas, the gut, and the brain at once.
The catch with the natural hormone is its half-life. Circulating GLP-1 is chewed up by an enzyme called dipeptidyl peptidase-4 (DPP-4) and cleared by the kidneys in 1 to 2 minutes, and only about 10 percent of what L-cells secrete ever reaches systemic circulation. [3] That tiny window is why the body's own GLP-1 cannot be repurposed as a drug. Pharma companies had to engineer molecules that resist DPP-4 and stick around for hours or days.
Two scientists are usually credited with mapping the system. Daniel Drucker (University of Toronto) and Jens Juul Holst (University of Copenhagen) characterized GLP-1's incretin role in the 1980s and early 1990s, work that opened the door to liraglutide, exenatide, and every later agent in the class. [3]
The four jobs GLP-1 does in the body
GLP-1 is part of a system called the incretin axis. When it binds the GLP-1 receptor (a G-protein-coupled receptor expressed on pancreatic islets, gut, brain, heart, and kidneys), it sets off four downstream effects that together explain why GLP-1 medications lower glucose and body weight. [1][3]

- Glucose-dependent insulin secretion. GLP-1 amplifies insulin output from pancreatic beta cells, but only when blood glucose is already elevated. That is why GLP-1 drugs rarely cause hypoglycemia on their own, in contrast to sulfonylureas or basal insulin. [2]
- Glucagon suppression. GLP-1 quiets alpha cells in the pancreas, lowering glucagon and reducing the liver's output of glucose between meals. The glucagonostatic effect is roughly as important as the insulin effect for overall A1C reduction. [3]
- Slowed gastric emptying. Food sits in the stomach longer, so glucose trickles into the bloodstream instead of spiking. This is also a major reason patients feel full faster and report nausea early in treatment. [1]
- Central appetite suppression. GLP-1 receptors in the hypothalamus and hindbrain register the signal as satiety. Patients commonly describe a quieter "food noise" and smaller portion sizes within the first few weeks. [3][6]
Inside the cell: how the GLP-1 receptor actually fires
The GLP-1 receptor (GLP-1R) is a class B G-protein-coupled receptor with seven transmembrane helices and a large extracellular domain that grabs the peptide. When GLP-1 (or a drug like semaglutide) docks, the receptor changes shape and recruits a Gαs heterotrimeric G-protein on the cytoplasmic side. [7] Gαs activates adenylyl cyclase, which converts ATP into cyclic AMP. Within seconds, intracellular cAMP rises sharply.
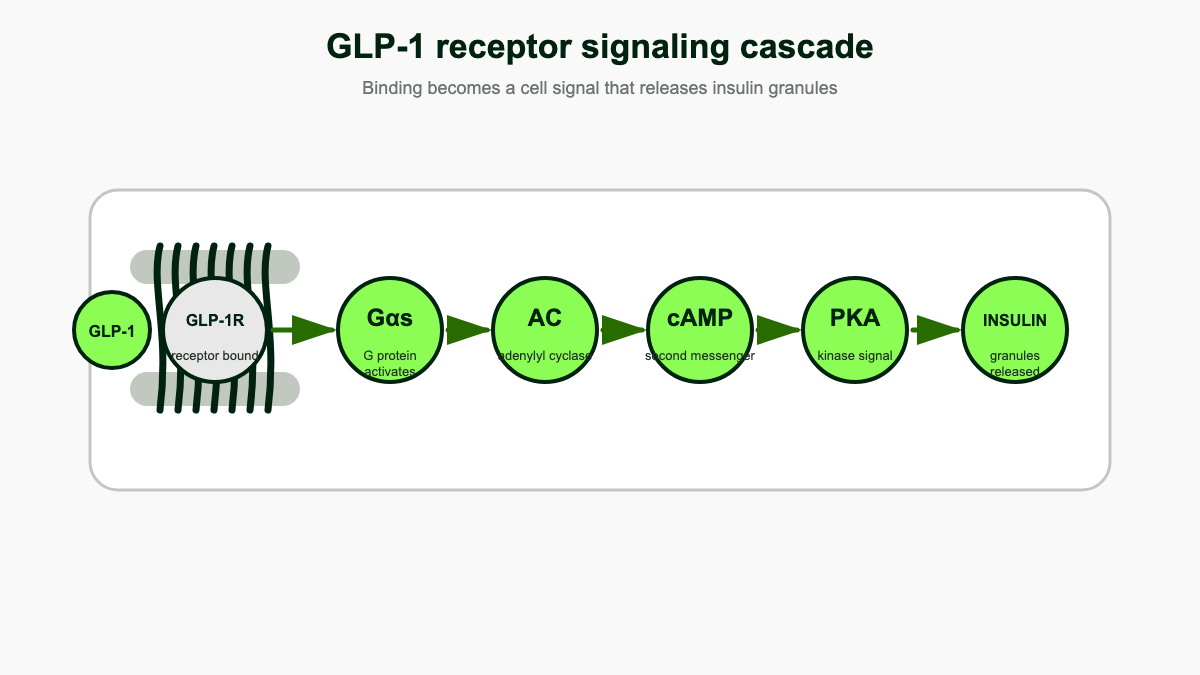
That cAMP pulse drives two parallel effector arms: protein kinase A (PKA) and a guanine nucleotide exchange factor called Epac2. In a pancreatic beta cell, both arms feed into the same outcome of glucose-dependent insulin release. [8]
The beta-cell pathway, step by step
When blood glucose is elevated, glucose enters the beta cell through GLUT2, gets phosphorylated by glucokinase, and feeds the citric acid cycle, raising the cell's ATP/ADP ratio. That rise closes ATP-sensitive potassium channels (the same KATP channels targeted by sulfonylureas), depolarizes the membrane, and opens voltage-gated L-type calcium channels. Calcium floods in and triggers fusion of insulin-loaded granules with the plasma membrane. [8]
GLP-1 amplifies every step downstream of the glucose signal. PKA phosphorylates the SUR1 subunit of KATP channels, making them more likely to close. Epac2 binds SUR1 in parallel, so even mice with disrupted PKA still respond to GLP-1 if Epac2 is intact. PKA and Epac2 also mobilize calcium from intracellular stores by activating IP3 and ryanodine receptors on the endoplasmic reticulum. Finally, both kinases phosphorylate vesicle-tethering proteins (synapsin-1, RIM2, Piccolo) that pull insulin granules closer to the membrane and ready them for fast release. [8]
The reason GLP-1 does not cause hypoglycemia on its own is that this entire amplification cascade only matters when glucose is already raising the ATP/ADP ratio. With normal or low glucose, the KATP channel stays open, the cell stays quiet, and the cAMP signal has nothing to amplify. [2]
Biased agonism and why molecule design matters
GLP-1R can also recruit beta-arrestins, which terminate signaling and pull the receptor inside the cell for degradation. Drugs that engage beta-arrestin strongly cause faster receptor desensitization. Drugs that favor the cAMP arm, like semaglutide and tirzepatide, keep the receptor signaling longer at the membrane. Tirzepatide in particular shows clear bias: a study in JCI Insight measured it as roughly 51 percent efficacious for cAMP at the GLP-1 receptor but less than 10 percent efficacious for beta-arrestin recruitment. [9] That kind of biased agonism is one reason newer molecules outperform native GLP-1 even when their receptor binding is similar.
How GLP-1 receptor agonists copy the hormone
GLP-1 receptor agonists are engineered peptides that bind the same receptor as native GLP-1 but resist DPP-4 breakdown. Two design tricks dominate the market.
Fatty-acid acylation (the semaglutide and liraglutide approach)
Liraglutide (Victoza, Saxenda) and semaglutide (Ozempic, Wegovy, Rybelsus) attach a fatty-acid chain to the peptide. The chain binds albumin in the bloodstream, which shields the molecule from DPP-4 and the kidneys. Liraglutide's half-life stretches to about 13 hours (once-daily dosing). Semaglutide's reaches roughly 165 hours, or about a week, which is what allows once-weekly Ozempic and Wegovy injections. [3]
Fc-fusion and exendin-based designs
Dulaglutide (Trulicity) fuses two modified GLP-1 peptides to an antibody Fc fragment, giving a 90-hour half-life. Exenatide (Byetta, Bydureon) is based on exendin-4, a peptide originally isolated from Gila monster venom that shares about 50 percent sequence with human GLP-1 but resists DPP-4 naturally. [3]
Tirzepatide: a dual GIP/GLP-1 agonist
Tirzepatide (Mounjaro for type 2 diabetes, Zepbound for obesity) is the first approved drug that activates both the GLP-1 receptor and the GIP (glucose-dependent insulinotropic polypeptide) receptor. [4] In a human pancreatic beta-cell line, tirzepatide produces a cAMP response that is significantly higher than GLP-1 or GIP alone, because the two receptor populations summate. [10] GIP also has effects GLP-1 lacks: it suppresses glucagon during high glucose but supports it during hypoglycemia (smoothing both ends of the glycemic range), and animal data suggest GIP signaling in hindbrain and hypothalamus blunts the nausea response normally driven by GLP-1 receptor activation in the area postrema. [10] Across head-to-head trials, tirzepatide produces roughly 5 percentage points more weight loss than semaglutide. [6]
Retatrutide and the triple agonist class
Retatrutide is an investigational once-weekly peptide that activates GLP-1, GIP, and glucagon receptors. Adding glucagon agonism is counterintuitive (glucagon raises glucose), but at low chronic exposure glucagon receptor activation in the liver appears to increase resting energy expenditure and fatty acid oxidation without driving hyperglycemia, because the GLP-1 component holds glucose in check. In a phase 2 trial published in NEJM in 2023, retatrutide produced about 24 percent body-weight loss at 48 weeks at the highest dose, the largest effect any pharmacologic agent for obesity has shown to date. [11] Phase 3 trials are ongoing.
Why the weight comes off
Weight loss with GLP-1 medications is not a side effect. It is a direct consequence of the mechanism. Three pathways drive it.
First, slowed gastric emptying means meals feel filling sooner and stay filling longer. Second, GLP-1 receptor agonists reach the brain and act on appetite circuits in the hypothalamus and brainstem, dialing down hunger signals at the source. Third, peripheral GLP-1 receptors on vagal afferent nerves carry satiety messages from the gut to the brainstem. [3]
There is also evidence from rodent and human imaging studies that semaglutide modifies dopamine-mediated food reward, which lines up with patient reports of reduced cravings for high-fat and high-sugar foods. [3][12]
The brain map: where GLP-1 quiets appetite
Long-acting GLP-1 drugs do not need to cross the entire blood-brain barrier to act on the brain. The hypothalamus and the dorsal vagal complex sit next to circumventricular organs (the median eminence and the area postrema) that have a leaky barrier on purpose, so peripheral hormones can reach central neurons. That access lets injected semaglutide and tirzepatide engage receptors in at least four anatomically distinct circuits.
Arcuate nucleus of the hypothalamus
The arcuate nucleus contains two opposing neuron populations: POMC neurons that suppress appetite by releasing alpha-MSH, and AgRP/NPY neurons that drive hunger. GLP-1 receptors are expressed on POMC neurons. Patch-clamp work in mouse brain slices shows that exendin-4 depolarizes POMC neurons and increases their firing rate, while indirectly silencing AgRP neurons through GABA inputs. [13] Activating arcuate GLP-1R neurons chemogenetically reduces food intake without changing glucose handling, which isolates appetite from glycemic effects. [13]
Nucleus tractus solitarius
The NTS in the brainstem receives vagal afferents from the gut and contains its own population of preproglucagon neurons that synthesize GLP-1 locally. NTS GLP-1R neurons project to the hypothalamus and the parabrachial nucleus and mediate the satiation that ends a meal. Knocking down GLP-1R selectively in vagal afferents increases meal size in mice, suggesting that physiological satiety relies on this gut-to-brainstem axis. [14]
Mesolimbic reward circuit
GLP-1 receptors are also expressed on dopamine neurons in the ventral tegmental area and in the nucleus accumbens. Activation in this circuit blunts the dopamine release triggered by palatable food, which maps onto the clinical observation that patients on semaglutide and tirzepatide report less interest in alcohol, sweets, and ultra-processed snacks. [12][14] The same circuit overlap is what has driven interest in GLP-1 agents for alcohol use disorder and other addictive behaviors.
Why "food noise" gets quieter
Patients often describe a drop in intrusive food thoughts within a few weeks of starting a GLP-1 drug. The likely substrate is the combined arcuate, NTS, and mesolimbic effect: hunger signals from AgRP neurons fall, satiation signals from the NTS rise sooner per gram of food eaten, and food-cued dopamine bursts are smaller. None of this is willpower. The circuits that normally dominate food-seeking are simply turned down.
Trial averages give a sense of the magnitude:
- Semaglutide 2.4 mg weekly (Wegovy): about 15 percent mean body-weight loss at 68 weeks in the STEP trials. [2]
- Tirzepatide 15 mg weekly (Zepbound): about 20 to 22 percent mean weight loss at 72 weeks in SURMOUNT-1. [2][6]
- Liraglutide 3.0 mg daily (Saxenda): about 5 to 8 percent at 56 weeks. [3]
- A1C reductions across the class generally fall between 0.8 and 1.8 percentage points in people with type 2 diabetes. [3]
The current GLP-1 medication lineup
All currently approved GLP-1 (and GLP-1/GIP) medications are peptides, which is why most are injected. Rybelsus is the one oral exception, made possible by a co-formulated absorption enhancer called SNAC. [3]
- Semaglutide: Ozempic (weekly injection, type 2 diabetes), Wegovy (weekly injection, obesity), Rybelsus (daily oral tablet, type 2 diabetes). [1][3]
- Tirzepatide: Mounjaro (weekly injection, type 2 diabetes), Zepbound (weekly injection, obesity and obstructive sleep apnea in adults with obesity). [4]
- Liraglutide: Victoza (daily injection, type 2 diabetes), Saxenda (daily injection, obesity). [1]
- Dulaglutide: Trulicity (weekly injection, type 2 diabetes). [1]
- Exenatide: Byetta (twice-daily injection) and Bydureon BCise (weekly injection), both for type 2 diabetes. [1]
Injections go into the abdomen, thigh, or upper arm and are designed for self-administration with prefilled pens.
Beyond glucose and weight: other effects of the same mechanism
Because GLP-1 receptors are present in the heart, blood vessels, kidneys, and immune cells, the drugs do more than just shrink the number on the scale.
A meta-analysis of seven cardiovascular outcome trials covering 42,920 patients with type 2 diabetes found a 12 percent relative reduction in major adverse cardiovascular events, a 12 percent reduction in all-cause mortality, and a 9 percent reduction in heart failure hospitalizations with GLP-1 receptor agonists. [3] Kidney composite endpoints fall by roughly 17 percent, mostly driven by less new-onset macroalbuminuria. [3]
Wegovy carries an FDA label for reducing the risk of cardiovascular death, heart attack, and stroke in adults with established cardiovascular disease and either obesity or overweight, based on the SELECT trial. [2]
Why the heart benefit is mostly not about weight loss
A subanalysis of SELECT presented in 2024 estimated that only about a third of the 20 percent reduction in major adverse cardiovascular events could be attributed to changes in waist circumference. [15] Patients across the entire baseline weight range got similar protection, and the magnitude of weight loss at 20 weeks did not predict who avoided cardiac events. The implication is that semaglutide acts directly on the cardiovascular system. Plausible mechanisms include lower vascular inflammation (high-sensitivity CRP drops markedly on semaglutide), improved endothelial nitric oxide signaling, modest reductions in systolic blood pressure, atherosclerotic plaque stabilization, and reduced platelet activation. GLP-1 receptors on macrophages and endothelial cells may directly damp the inflammatory milieu that drives plaque rupture. [3][15]
Kidney protection and the FLOW trial
The FLOW trial, published in NEJM in 2024, randomized 3,533 adults with type 2 diabetes and chronic kidney disease to once-weekly semaglutide 1.0 mg or placebo. The trial was stopped early for efficacy. Semaglutide cut a composite of major kidney outcomes (kidney failure, sustained 50 percent eGFR loss, or kidney/cardiovascular death) by 24 percent. [16] Mechanism analyses suggest only part of the effect comes from weight, blood pressure, and HbA1c improvements. Direct kidney mechanisms include suppression of inflammatory cytokines, reduced oxidative stress, and natriuresis through enhanced atrial natriuretic peptide release and reduced sodium reabsorption in the proximal tubule. [16][17] Benefit holds in patients already on SGLT2 inhibitors, suggesting the two classes work through complementary pathways.
Side effects tied directly to the mechanism
The most common side effects are gastrointestinal, and they make sense given how the drug works. Slowing the stomach and reducing appetite produces nausea, vomiting, diarrhea, constipation, and reflux, especially in the first several weeks and after each dose escalation. [1][3]
Why the nausea: the area postrema
The area postrema is a small region in the brainstem that lacks a normal blood-brain barrier. It is the brain's chemical sensor for circulating toxins, which is why drugs as different as chemotherapy and apomorphine can trigger nausea through it. The area postrema also contains GLP-1 receptors. Patch-clamp experiments show that GLP-1 directly excites about half of area postrema neurons via the cAMP cascade, and that excitation propagates to the nucleus tractus solitarius and downstream emetic circuitry. [18] When you titrate a long-acting GLP-1 agonist up too quickly, you saturate area postrema receptors faster than the rest of the central nervous system can adapt, and nausea results. Slow titration lets the area postrema receptors partially desensitize before peripheral signaling reaches steady state.
Most GI symptoms are mild to moderate and ease with time, slower titration, smaller meals, and avoiding fatty or fried foods. Less common but more serious risks include acute pancreatitis, gallbladder disease (cholelithiasis occurs in roughly 1 to 3 percent of patients depending on agent and dose), acute kidney injury from dehydration, and in people with preexisting type 2 diabetes, transient worsening of diabetic retinopathy. [3]
GLP-1 medications carry a boxed warning against use in people with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2, based on rodent studies. The signal has not been confirmed in humans, but the contraindication remains. [1][5]
Why titration matters: receptor desensitization
Every GLP-1 product label calls for slow dose escalation, typically over 16 to 20 weeks for the obesity doses. The biological reason is twofold. First, the area postrema adaptation described above. Second, GLP-1R itself desensitizes when activated continuously: beta-arrestins recruit kinases that uncouple the receptor from Gαs and pull it inside the cell. Starting low and stepping up gives receptors time to internalize and recycle in a stable rhythm rather than getting hammered all at once, which improves both tolerability and the eventual maximum effect. Patients who drop the dose during a flare and then re-escalate usually recover the full benefit, because receptor expression returns once exposure stabilizes. [3]
Type 2 diabetes versus obesity: same drug, different center of gravity
The same molecule (semaglutide) is sold as Ozempic for diabetes and Wegovy for obesity, with the only difference being the maximum dose (2.0 mg weekly versus 2.4 mg weekly). Tirzepatide is similarly split between Mounjaro and Zepbound. The mechanism does not change, but which arm of the mechanism dominates the clinical benefit does.
In type 2 diabetes, glucose-dependent insulin secretion and glucagon suppression do most of the work. A1C drops by 1 to 2 percentage points within the first 12 to 16 weeks, and that effect plateaus relatively early because beta-cell function and glucagon tone normalize. [3] In obesity without diabetes, the same insulin and glucagon effects are present but smaller, and the dominant signal is central appetite suppression. Weight loss continues for 60 to 72 weeks before plateauing in the STEP and SURMOUNT trials, mirroring the slower neuroplastic changes in hypothalamic and reward circuits. [2][6]
This is also why the highest tolerated doses are reserved for obesity. Pushing past the dose where glycemic effects max out keeps adding receptor occupancy in the brain, where the dose-response for appetite is shallower and runs longer.
Why some people don't respond
About 10 to 15 percent of people in clinical trials lose less than 5 percent of body weight on a maximum-dose GLP-1 agonist. Several biological explanations are converging.
A 2026 Stanford-led genome-wide study identified two missense variants in the PAM gene (encoding peptidyl-glycine alpha-amidating monooxygenase, an enzyme that adds the C-terminal amide that makes GLP-1 active) that predict poor response. Carriers of the p.S539W variant achieved blood-sugar targets at less than half the rate of non-carriers in a meta-analysis of more than 1,100 patients. Carriers actually have higher circulating GLP-1, but the hormone seems less effective downstream, a phenomenon the researchers compare to insulin resistance. [19]
Other contributors to non-response include rapid receptor desensitization in some patients, GIP receptor variation (relevant for tirzepatide), differences in vagal afferent density, and behavioral factors like compensatory snacking on liquid calories that bypass the gastric-emptying brake. Switching agents (for example, tirzepatide after semaglutide failure) helps a meaningful subset, suggesting receptor-level differences matter even within the class. [19]
How Rybelsus works: getting a peptide through the stomach
Peptides do not normally survive the stomach. They are denatured by acid and chopped up by proteases before they can cross the intestinal wall. Rybelsus is the only oral GLP-1 currently on the market, and it works because each tablet co-formulates semaglutide with a small molecule called sodium N-(8-(2-hydroxybenzoyl)amino)caprylate, or SNAC. [20]
SNAC does three things in the immediate vicinity of the tablet as it sits against the stomach wall. It buffers the local pH so semaglutide is not destroyed by acid. It prevents semaglutide molecules from clumping into the inactive aggregates that block absorption. And it transiently increases the fluidity of the gastric epithelial membrane so the peptide can cross. The window of opportunity is narrow: bioavailability is about 0.8 percent compared with the injection. [20]
That is why Rybelsus has strict dosing rules: take with no more than 4 ounces of plain water, on an empty stomach, and wait at least 30 minutes before any food or other oral medication. More than half of patients in the dose-finding studies who took it with food had no measurable drug exposure at all. [20] The molecule itself has the same week-long half-life as injected semaglutide; the daily dosing is a workaround for the low and variable absorption.
What happens when you stop: the regain biology
GLP-1 medications work as long as the drug is on board. When patients stop, the effect goes away. The STEP-1 extension trial showed participants regained roughly two-thirds of their lost weight within a year of stopping semaglutide. [2] A 2025 BMJ analysis pooling withdrawal data across multiple GLP-1 agents found similar trajectories: most of the lost weight returns within 12 to 24 months. [21]
The biology is straightforward. Body weight in humans is defended by overlapping hormonal and neural set-point mechanisms. Weight loss of any kind (caloric restriction, surgery, GLP-1 drugs) lowers leptin, raises ghrelin, slows resting metabolic rate slightly, and increases hunger drive. While the drug is working, the appetite-suppressing GLP-1 signal overrides those defenses. When the drug clears, the defenses are still there. [21] Researchers at the University of Alabama at Birmingham reported in 2025 that residual leptin resistance after GLP-1-induced loss may keep the satiety signal from re-engaging properly, which explains why the rebound is fast rather than gradual. [22]
The practical implication, recognized by every major clinical guideline, is that obesity treated with GLP-1 medications functions like hypertension treated with antihypertensives: the disease is managed, not cured, and stopping treatment usually means losing the benefit. Strategies under investigation to extend the effect include resistance training during loss to preserve lean mass, gradual dose tapering rather than abrupt stops, and combining GLP-1 with leptin sensitizers or agents that target alternative pathways.
What the mechanism does not do
GLP-1 drugs (with the partial exception of glucagon-co-agonists like retatrutide) do not "burn fat" or raise resting metabolic rate. Weight loss happens because patients eat less, not because energy expenditure increases. That has two practical implications.
First, weight regain after stopping treatment is common, for the reasons covered in the previous section. Second, muscle loss accompanies fat loss, as it does in any caloric deficit. Body composition studies of semaglutide and tirzepatide suggest that 25 to 40 percent of the weight lost is lean mass. Resistance training, adequate protein intake (roughly 1.2 to 1.6 grams per kilogram of reference body weight per day), and slower titration help preserve muscle. None of this is unique to GLP-1 agents; it applies to any sustained energy deficit.
GLP-1 drugs also do not directly fix insulin resistance, the underlying defect in type 2 diabetes. They lower glucose by amplifying the insulin-secretion response from beta cells that still work. In long-standing diabetes where beta-cell mass has dropped substantially, the glycemic response is smaller, and combination therapy (with metformin, an SGLT2 inhibitor, or basal insulin) is usually still needed. [3]
References
- Cleveland Clinic. GLP-1 Agonists: What They Are, How They Work & Side Effects. Cleveland Clinic, 2024. my.clevelandclinic.org
- Harvard Health Publishing. How does Ozempic work? Understanding GLP-1s for diabetes, weight loss, and beyond. Harvard Medical School, 2024. health.harvard.edu
- Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes: state-of-the-art. Molecular Metabolism, 2021. PMC8346189. pmc.ncbi.nlm.nih.gov
- U.S. Food and Drug Administration. FDA approves new medication for chronic weight management (tirzepatide / Zepbound). FDA News Release, 2023. fda.gov
- U.S. Food and Drug Administration. Highlights of Prescribing Information: Ozempic (semaglutide) injection. Novo Nordisk / FDA, 2024. accessdata.fda.gov
- WMCHealth. Beyond the Ozempic Buzz: How GLP-1s Actually Work. Westchester Medical Center Health Network, 2024. wmchealth.org
- Zhang Y, Chen S, Wang Z, et al. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduction and Targeted Therapy (Nature), 2024. nature.com
- Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas: cAMP, PKA, Epac2, and insulin granule exocytosis. PMC3118608. pmc.ncbi.nlm.nih.gov
- Willard FS, Douros JD, Gabe MBN, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight, 2020. insight.jci.org
- Min T, Bain SC. The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes. PMC, 2021. pmc.ncbi.nlm.nih.gov
- Jastreboff AM, Kaplan LM, Frias JP, et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity (Phase 2). New England Journal of Medicine, 2023. nejm.org
- Volkow ND, Wang GJ. GLP-1 receptor agonists, dopamine, and food reward. Reviewed in Endocrinology, 2025. academic.oup.com
- Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. Journal of Clinical Investigation, 2014. gubra.dk
- Brierley DI, Holt MK, Singh A, et al. GLP-1 and the neurobiology of eating control: NTS, vagal afferents, and reward circuits. Endocrinology, 2025. academic.oup.com
- Deanfield J, Verma S, Scirica BM, et al. SELECT: Semaglutide and cardiovascular outcomes by baseline weight (subanalysis). Reported via TCTMD, 2024. tctmd.com
- Perkovic V, Tuttle KR, Rossing P, et al. Effects of Semaglutide on Chronic Kidney Disease in Patients with Type 2 Diabetes (FLOW). New England Journal of Medicine, 2024. nejm.org
- Mann JFE, Rossing P, Bakris G, et al. FLOW trial design and rationale. Nephrology Dialysis Transplantation, 2023. PMC10469096. pmc.ncbi.nlm.nih.gov
- Yamamoto K, Iwasaki Y, Maejima Y, et al. Glucagon-like peptide-1 (GLP-1) action in the mouse area postrema neurons. Peptides, 2018. PubMed 30081042. pubmed.ncbi.nlm.nih.gov
- Stanford Medicine. One in 10 people may have resistance to GLP-1 diabetes drugs (PAM gene variants). Stanford News, 2026. med.stanford.edu
- Aroda VR, Blonde L, Pratley RE. The Oral Semaglutide Experience: SNAC technology and clinical practice. PMC10788673, 2024. pmc.ncbi.nlm.nih.gov
- BMJ Editorial Team. Weight regain after cessation of medication for obesity. BMJ, 2025. bmj.com
- University of Alabama at Birmingham. New UAB discovery on weight regain after stopping GLP-1 treatment (leptin sensitivity). UAB News, 2025. uab.edu